
【核心研究背景】
过氧化氢(H2O2)作为世界上100种最重要的化学品之一,广泛应用于废水处理、医疗消毒及化学合成。然而,传统蒽醌法能耗高、污染重,氢氧直接合成法存在爆炸风险。更关键的是,H2O2的远程运输不仅成本高,且存在安全隐患。利用分子氧(O2)和水(H2O)的光催化合成H2O2方法,为可持续和经济的H2O2合成提供了一条有吸引力的途径。与下游催化反应相结合的光生 H2O2原位利用系统,可以实现光生H2O2的直接利用,有效避免了H2O2的分离和运输。尽管前景广阔,但受电荷分离性能差,O2、质子(H+)供应不足等因素制约,目前报道的光催化效率普遍较低。
【催化剂的制备与关键表征】
本篇博士学位论文为实现光催化H2O2的高效生成与原位利用,以有机聚合物为平台,通过官能团设计,系统性强化了电子、O₂和H⁺的供应,并在此基础上实现了光生H₂O₂的原位高效利用。
为强化电子供应,采用希夫碱缩合反应合成了二苯甲酮功能化聚合物(COP-BPN)。FT-IR、13C NMR以及XPS证实了C=O的存在。N2吸脱附表明所制备的聚合物均为无定形网络且比表面积相近,排除了形貌对催化性能的干扰,为后续性能差异归因于电子结构奠定了基础。为解决O2和H⁺供应不足的问题,以SBA-15为载体,采用气相辅助固相合成法,在三氟甲烷磺酸蒸气催化下通过氰基三聚反应制备三联苯功能化共价三嗪聚合物(CTP-TD)。FT-IR(1506、1369 cm-1)和13C NMR(172 ppm)证实了三嗪环特征信号的出现。为实现H2O2原位利用于丙烯环氧化,将三聚氰胺-三聚氰酸超分子自组装前驱体在氩气中550℃热解制备了N3C空位修饰的氮化碳(CN-Ar)。XPS定量分析表明,表面N/C比从1.3498降至1.1686,N2C/N3C比值从1.83升至3.67,证实N3C空位的形成。最后,为深度活化H2O2转化为Cl2•−活化C-H键,以噻吩-2,5-二腈为单体,同样采用气相辅助固相合成法合成了CTP-S。FT-IR和13C NMR证实了三嗪环的形成及噻吩结构的保留(噻吩碳信号在132、147 ppm)。
【光催化性能与机理】
论文围绕强化反应物供应(电子、O2、H⁺)→高效光合成H2O2→原位转化利用这一主线,设计并验证了四种具有代表性的有机聚合物光催化体系。每个体系不仅在性能上取得了突破,更在反应机理上实现了创新。
(1)二苯甲酮功能化聚合物(COP-BPN):拓展共轭,强化电子供应
在纯水+O2、无任何牺牲剂的条件下,COP-BPN的H2O2生成速率高达3527 μmol·g⁻¹·h⁻¹,是对照组的4.5倍。机理研究表明,二苯甲酮结构的引入不仅可以通过拓展共轭结构加速光生电荷的分离和转移效率,而且能够显著降低O2吸附能,这些特性有助于高效光催化生成H2O2。
(2)三联苯功能化三嗪聚合物(CTP-TD):调控能量转移,强化O2与H⁺双供应
在无额外牺牲试剂的条件下,CTP-TD显示出优异的光生H2O2性能(1342 μmol·g-1·h-1,2013 μM·h-1)。实验和理论研究表明,与联苯结构相比,三联苯结构的引入显著改善了O2吸附和水氧化过程。三联苯结构通过促进系间窜越(ISC)过程提高单线态氧(1O2)产量。亲电性的1O2比O2更容易吸附在催化剂表面,有利于后续的氧还原过程。此外,三联苯结构上积累的空穴可以降低水氧化能垒,增强了H2O2形成过程中的质子供应。
(3)氮空位修饰氮化碳(CN-Ar):原位利用光生H2O2高选择性制备环氧丙烷(PO)
为了满足HPPO的要求(防止环氧丙烷开环,同时有效获得H2O2),利用氩气热解和超分子自组装前驱体的协同作用,一步制得具有N3C空位的氮化碳用于高效光合成H2O2。在光照和O2环境下,串联体系可以高效(5515 μmol·g-1·h-1)和高选择性(99.1%)的生产环氧丙烷(PO)。机理研究表明,N3C空位的引入显著促进了光生电子空穴分离,增强了H+和O2的吸附。涉及•O2−和1O2的双合成路径促进了H2O2的高效生成(5775 μmol·g-1·h-1,5775 μM·h-1)。此外,兼容的溶剂体系(甲醇/水)同时促进了H2O2和PO的生成,有效避免了H2O2的分离纯化。
(4)噻吩基三嗪聚合物(CTP-S:深度活化O2为Cl2•−,打破氯化动力学限制
供体(噻吩)-受体(三嗪)结构赋予了CTP-S优异的光生电子-空穴分离能力,深度活化O2为Cl2•−(O2→H2O2→•OH→Cl•→Cl2•−)。在酸性-两相一体化体系(2 M HCl水相/甲苯有机相)中,甲苯转化速率达到16.94 mmol·g⁻¹·h⁻¹,苯甲醛选择性99.5%。闪光光解证实Cl2•−在340 nm处有特征吸收,寿命长达33.34 μs(远长于Cl•的<5 μs和•OH的~100 ns)。与Cl•相比,长寿命的Cl2•−能够更有效地连续活化C(sp3)-H生成不稳定的二氯中间产物,将二氯化与一氯化的动力学速率比提升2000倍,打破了传统二氯化动力学限制。这些活性中间体比稳定的二氯化合物更容易水解成醛类或酮类,有效避免了氯化副产物的产生。此外,酸性-两相一体化体系强化了Cl2•−介导的过程,抑制了产物过氧化。该体系对环己烷、乙苯、对二甲苯等多种底物均表现出高转化率和高醛/酮选择性,且无一氯代副产物。这是首次将Cl2•−应用于惰性C(sp3)-H键选择性制备醛/酮,为光生H2O2的深度原位利用开辟了新方向。
【图文速览】
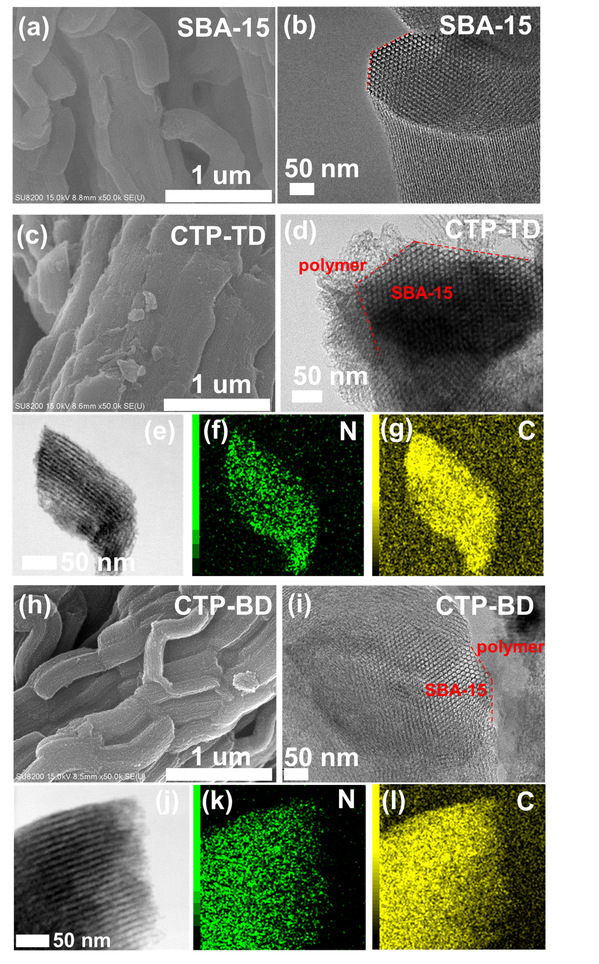
图1 所制备催化剂(CTP-TD)的微观形貌
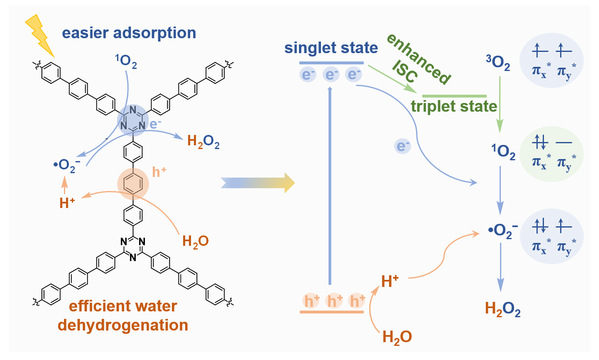
图2 CTP-TD光催化产H2O2机理
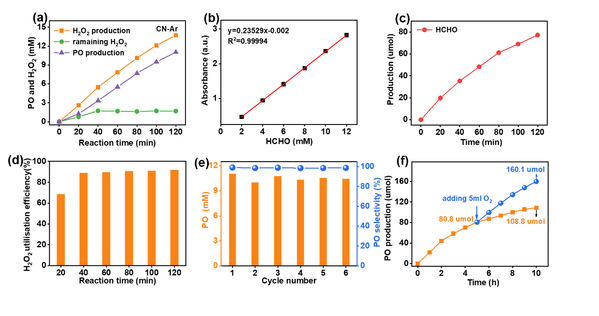
图3 CN-Ar光催化产H2O2串联丙烯环氧化体系的催化性能
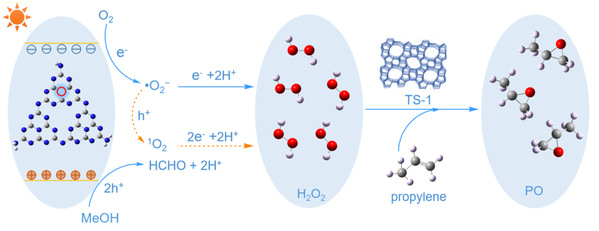
图4 CN-Ar光催化合成PO的反应机理图
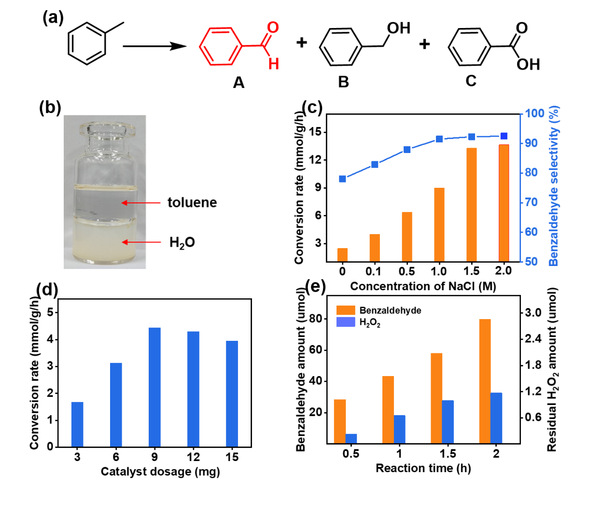
图5 CTP-S光催化甲苯氧化制备苯甲醛性能
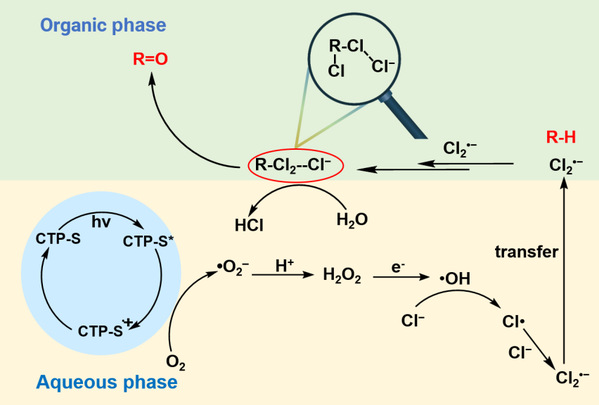
图6 CTP-S光催化甲苯氧化机理
【研究结论】
本篇优秀学位论文以有机聚合物为出发点,利用官能团功能化选择性调控电子转移和能量转移过程,证实了强化电子(e−)、O2和H+供应是实现H2O2高效生成的有效策略。在此策略指导下进一步设计适用于原位利用系统的高效光催化剂,实现了光生H2O2的直接原位利用和深度转化利用。
